
This post has been republished via RSS; it originally appeared at: Microsoft Research.
While there has been a focus in the quantum computing industry on growing the number of qubits in a quantum computer, the reality is there are many important factors when building an overall system to bring quantum solutions to fruition. Hardware scaling, temperature control, software optimizations, and many other considerations must be reimagined in ways that allow large-scale quantum computers to do the necessary, meaningful work to solve some of today’s and tomorrow’s biggest problems. A question emerges that is both scientific and philosophical in nature: once a quantum computer scales to handle problems that classical computers cannot, what problems should we solve on it? Quantum researchers at Microsoft are not only thinking about this question—we are producing tangible results that will shape how large-scale quantum computer applications will accomplish these tasks.
We have begun creating quantum computer applications in chemistry, and they could help to address one of the world’s biggest challenges to date: climate change. In January, Microsoft launched a bold new environmental sustainability initiative focusing on carbon, water, waste, and biodiversity, announcing one of the most ambitious carbon commitments put forward by any company: Microsoft will be carbon negative by 2030 and remove from the environment more carbon than we have emitted since our founding by 2050. Last week, we announced seven important new steps on our path to be carbon negative by 2030. Learn more on the Microsoft on the Issues blog.
Microsoft has prioritized making an impact on this global issue, and Microsoft Quantum researchers have teamed up with researchers at ETH Zurich to develop a new quantum algorithm to simulate catalytic processes. In the context of climate change, one goal will be to find an efficient catalyst for carbon fixation—a process that reduces carbon dioxide by turning it into valuable chemicals. One of our key findings is that the resource requirements to implement our algorithm on a fault-tolerant quantum computer are more than 10 times lower than recent state-of-the-art algorithms. These improvements significantly decrease the time it will take a quantum computer to do extremely challenging computations in this area of chemistry. In our research, we have not only improved upon quantum algorithms and have shown how they can help effectively find new catalysts, we have also learned more about other quantum resources that are necessary to perform these calculations at an exponentially faster rate than classical computers. These learnings include the size of quantum computers and their runtime—and more generally how to better co-design a hybrid quantum-classical computing system to handle this type of problem. Our research is detailed in a paper called “Quantum computing enhanced computational catalysis.”
Carbon fixation: An opportunity in chemistry opens the door for a new application in quantum computing

Synthetic carbon fixation is a process that has potential to help greatly reduce carbon dioxide in the atmosphere by converting CO2 into other useful chemical compounds. Carbon fixation is not a new process. In fact, it is a very old one. Plants use a form of carbon fixation to convert carbon dioxide into energy-rich molecules such as glucose. But glucose isn’t the only possible biproduct of carbon fixation. When using different catalysts, natural or synthetic, carbon dioxide can be converted into other compounds.
Currently, synthetic catalytic processes are found through lengthy trial-and-error lab experiments. In a process that requires testing thousands of molecular combinations, computer simulations that very accurately model quantum correlations could replace complex synthesis of new candidate catalysts. Whereas computers today can have a difficult time accurately calculating properties of complex molecules, quantum computers are especially suited for this task and will give more reliable and predictive simulation results. We hope that quantum computers will complement traditional methods and, together, could reveal a process that both removes carbon dioxide from the atmosphere and provides valuable chemicals in return.
| Why begin with a known catalytic reaction if the goal is to find new ones? |
In order to better understand how quantum computer algorithms can assist in discovering new, more efficient catalysts, we decided to focus our analysis on a previously published catalytic process based on the transition metal Ruthenium to convert carbon dioxide into methanol. It is also—like all known catalysts resulting in methanol to date—extremely inefficient. This inefficiency offers an opportunity for finding catalytic reactions that are more scalable. Using this reaction as a foundation for testing our algorithm, we were able to gain knowledge about how to best optimize algorithms for simulating these types of reactions on a quantum computer (see Figure 1 above).
Our algorithmic advancement: Boosting computational efficiency through compression
We need to develop more efficient algorithms for quantum computers because problems that involve calculating molecular energies with high precision, such as for catalytic processes, will be resource intensive—even on quantum computers.
- ENGAGE Quantum Development Kit Are you a researcher or developer who wants to help in discovering new algorithms for quantum computers? Check out the Quantum Development Kit and Q#, a toolkit and high-level programming language for developing quantum algorithms, which allow you to try out a small chemical algorithm for yourself.
Obtaining high-precision energy estimates requires simulating the molecule’s quantum state for a long period of time, which is split into multiple smaller time steps. All the interaction terms in the problem description, the so-called Hamiltonian, need to be loaded over and over again at every single time step since quantum information cannot be copied. The natural approach to reduce overall runtime is then to reduce both the information that needs to be loaded as well as the number of time steps required for the simulation. One promising approach is to use a so-called “double-factorized” representation of the Hamiltonian. In this representation, the information describing the interaction between electrons is compressed into fewer terms.
In our research, we precisely achieve this runtime reduction by developing a new, efficient quantum algorithm. Our algorithm exploits the improved compression properties of the double-factorized form, and it also manages to perform the simulation with significantly larger step sizes compared to prior state of the art that exploits the unfactorized or single-factorized forms of the Hamiltonian. The extent of our improvement for molecules such as Ruthenium catalysts is driven primarily by the larger time step size, as illustrated in the table below. Moreover, aggressive compression can further reduce the number of terms at the cost of accuracy in the simulation. Importantly, our use of a so-called “qubitization” simulation algorithm allows for good control over the target accuracy. Combined, these factors reduce runtime by orders of magnitude for obtaining reliable results.
| Ruthenium catalyst configuration VIII with 130 spin-orbitals | ||
|---|---|---|
| Approach | Number of steps per unit of time evolution | Overall algorithmic speedup |
| Unfactorized | 10,600 | 1.0x |
| Single-factorized | 42,200 | 0.4x |
| Our results | 570 | 18.9x |
Designing quantum computers for the hunt for new catalysts
The computational design of catalysts relies on very accurate energy calculations. Quantum computers can avoid uncontrolled approximations of classical simulations. They scale much better and open opportunities to assess the energetics of chemical species with sufficient accuracy. By using the Hamiltonian parameters generated on classical computers, a quantum computer could solve the exact energies of the chemical systems and help profile a quantitatively accurate landscape of reaction pathways. Catalyst structures could then be further refined or modified through the insights generated by reaction kinetics analysis. Such a process could iterate until a desired catalyst is found.
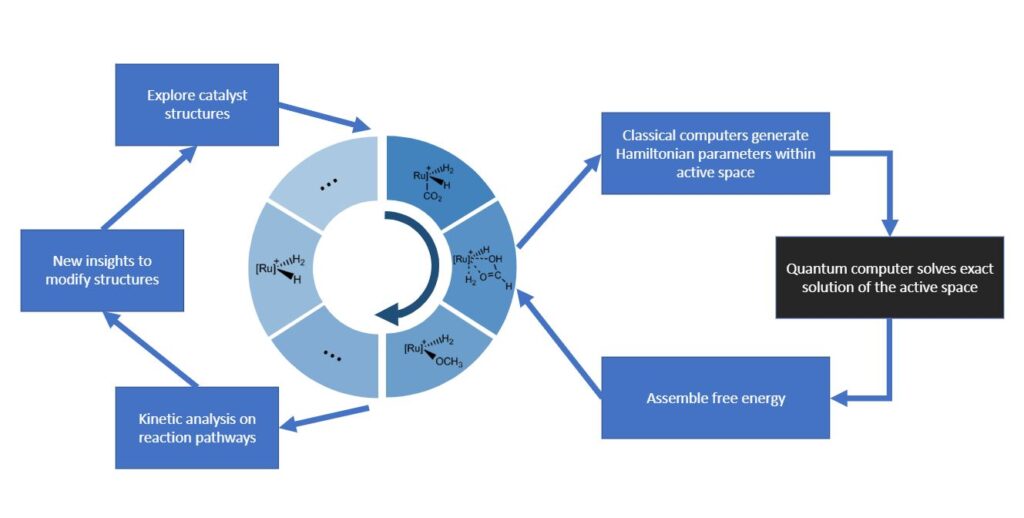
Our paper is the first to show analysis of a quantum algorithm being done on a specific chemical reaction along the entire reaction pathway. Instead of just a single configuration, we analyzed relevant configurations of the reactants along this pathway. In addition, we performed state-of-the-art classical calculations, but our results with these confirmed that they lack reliability for truly predictive computational catalysis. Thus, one of the first roles for quantum computers will be not only to provide accurate results for novel catalysts, but also to benchmark validity of various classical approximations and develop better classical simulation methods.
Beyond this, we want to further optimize quantum algorithms to enable the simulation of larger numbers of electrons. Current algorithms limit the accurate quantum computation to so-called active spaces of the most correlated electrons. While that may often be sufficiently accurate, we will not know unless we can simulate larger active spaces or ideally all electrons in a molecule.
Finally, with the estimates for gate counts calculated, we were able to translate this information into potential runtime estimates for quantum computation on this problem. Depending on the assumptions made about future quantum computers, we estimate that it may take anywhere from a little over a day to several years to perform such calculations. This clearly shows the need not only for fast algorithms but also fast and scalable quantum hardware.
Our newer, faster quantum algorithm for calculating molecular energy levels is itself an exciting development and a crucial step in the computational catalysis workflow (see Figure 2 above), but it will take more than that to find an efficient catalyst. In fact, knowing more about the quantum algorithms needed to undertake improved computational catalysis opens the door to even more questions about the scale of quantum computers. What is the amount of memory we need to run these algorithms at a meaningful speed? What does this imply for the needed hybrid workflow and quantum architecture it runs on to successfully find these catalysts? Our results after testing this algorithm reveal some important discoveries going forward.
Where do quantum computers and chemistry applications go from here?
The research presented in this post is evidence that rapid advances in quantum computing are happening now—our algorithm is 10,000 times faster than the one we created just three years ago. By gaining more insight into how quantum computers can improve computational catalysis, including ways that will help to address climate change while creating other benefits, we hope to spur new ideas and developments on the road to creating some of the first applications for large-scale quantum computers of the future. The advancements in algorithms and knowledge gained from our research are a springboard for future work, including exploring additional ways algorithms can be made even more effective. Given the promise and potential that quantum computing represents for tackling the toughest challenges in chemistry, we hope to work alongside the chemistry community to better understand how quantum computers can be best utilized to further develop new chemical processes, molecules, and, eventually, materials.
We are encouraging those who are interested in exploring how chemistry can be impacted by quantum computing to explore Azure Quantum, which comprises a full set of tools, ranging from the Quantum Development Kit (QDK) and the Q# programming language for quantum to simulators and resource estimators. The QDK allows researchers to develop and test new quantum algorithms for chemistry, run small examples on a simulator, use Azure Quantum on quantum hardware, and estimate resource requirements to run simulations at scale on future quantum computers.
As part of the QDK, we developed a Q# chemistry library, with our partner Pacific Northwest National Laboratories (PNNL), that provides several fundamental data structures and tools to explore quantum algorithms for chemistry. If you are looking to get started with the QDK and Q#, check out our Microsoft Learn modules released at Microsoft Build 2020.
The post State-of-the-art algorithm accelerates path for quantum computers to address climate change appeared first on Microsoft Research.